
Galactosemia is a rare and inherited metabolic disorder that affects the body’s ability to break down galactose, a sugar found in milk and other dairy products. It occurs when there is a deficiency of certain enzymes needed to convert galactose into glucose, resulting in the accumulation of toxic substances in the body.
Galactosemia is typically diagnosed in infancy through newborn screening or clinical symptoms, which can include vomiting, jaundice, poor feeding, and developmental delays. The condition requires strict dietary management, with the elimination of all sources of galactose from the diet, including breast milk and regular formula.
What is Galactosemia?
Hereditary galactosemia is among the most common carbohydrate metabolism disorders and can be a life-threatening illness during the newborn period.
- Galactosemia is a recessive hereditary metabolic disorder in which the enzyme necessary to convert galactose into glucose is missing.
- First described in a variant patient in 1935 by Mason and Turner, galactose-1-phosphate uridyltransferase (GALT) deficiency is the most common enzyme deficiency that causes hypergalactosemia.
- Removing lactose largely eliminates the toxicity associated with newborn disease, but long-term complications routinely occur, as reported by Komrower and Lee in 1970 and then delineated in a 1990 retrospective survey by Waggoner and associates.
Pathophysiology
Hypergalactosemia is associated with the following 3 enzyme deficiencies.
- Galactokinase converts galactose to galactose-1-phosphate and is not a common deficiency.
- Uridine diphosphate (UDP) galactose-4-epimerase epimerizes UDP galactose to UDP glucose and is also uncommon.
- GALT is responsible for hereditary galactosemia and is the most common deficiency. This enzyme catalyzes the conversion of galactose-1-phosphate and UDP glucose to UDP galactose and glucose-1-phosphate. Individuals with GALT deficiency manifest abnormal galactose tolerance.
Statistics and Incidences
Incidence is approximately 1 case per 40,000-60,000 persons.
- Incidence widely varies (ie, 1 case in 70,000 people in the UK but 1 case in 16,476 people in Ireland.); the disorder is thought to be much less common in Asians.
- Most patients appear to reach adulthood following the institution of a galactose-restricted diet.
- Galactosemia occurs in all races; however, galactosemia variants are based on the exact gene defect.
- Galactosemia equally affects males and females.
- Galactosemia is most often diagnosed in infancy by newborn screening because all states include galactosemia as part of their newborn screening.
Clinical Manifestations
Untreated infants with severely deficient galactose-1-phosphate uridyltransferase (GALT) activity typically present with the following variable findings:
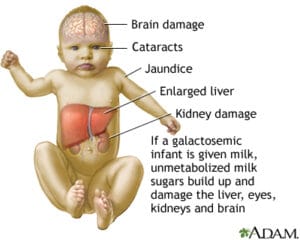
- Poor growth. Poor growth within the first few weeks of life of the infant.
- Jaundice. There is a yellowish discoloration of the infant’s skin because of hyperbilirubinemia.
- Bleeding. Bleeding from coagulopathy is also a symptom of galactosemia.
- Feeding difficulties. Early feeding difficulties with vomiting and diarrhea severe enough to produce dehydration and weight loss and jaundice are primary manifestations.
- Cataracts. Cataracts may occur unless milk is withheld early.
- Liver and spleen damage. The liver and spleen may dysfunction, leading to hepatomegaly and splenomegaly.
Assessment and Diagnostic Findings
Galactosemia can be identified earlier through newborn screening.
- Beutler test. A screening test called the Beutler test can be used to test for the disorder.
- Newborn screening. A positive (ie, abnormal) indication on the newborn screen must be followed by a quantitative erythrocyte galactose-1-phosphate uridyltransferase (GALT) analysis by a laboratory that routinely performs biochemical genetic testing and consultation.
- GALT isoelectric-focusing electrophoresis test. A GALT isoelectric-focusing electrophoresis test helps distinguish variant forms such as the Duarte defect.
- GALT genotyping. GALT genotyping may provide a specific molecular diagnosis; the most common GALT allele in Caucasians is the Q188R mutation. The S135L mutation is common in native South Africans and in African Americans.
Medical Management
The mainstay of medical care in the postnatal period is to immediately discontinue ingestion of lactose-containing formula.
- Transfusions. Clotting abnormalities may be cryptic and require fresh frozen plasma treatments.
- Diet. Prescribe a galactose-restricted diet for infants who are galactosemic; a substitution for milk, such as Nutramigen and Pregestimil, can provide galactose-free nutrition for the infants.
Pharmacologic Management
Drug therapy currently is not a component of the standard of care for this condition.
Nursing Management
Nursing care mainly centers on the intake of the child.
Nursing Assessment
Assessment of a child with galactosemia includes:
- Physical examination. Assess the child’s presenting symptoms, especially after ingestion of galactose.
- Nutritional intake. Assess the child’s dietary needs, and the family caregivers‘ understanding of the disorder to establish a strict diet regime.
Nursing Diagnosis
Based on the assessment data, the major nursing diagnoses are:
- Altered nutrition: less than body requirements related to a restrictive diet.
- Ineffective breastfeeding related to intolerance to galactose.
- Diarrhea related to ingestion of galactose-containing substance.
Nursing Care Planning and Goals
The major nursing care planning goals for a child with galactosemia are:
- The caregivers will be able to identify the appropriate food for the infant.
- The caregivers will be able to provide galactose-free milk as a substitution for breast milk.
- The caregivers will be able to understand the disease process and the care of the newborn with galactosemia.
Nursing Interventions
Nursing interventions for a child with galactosemia include:
- Milk substitution. A soy-based formula, meat-based formula, Nutramigen, or another soy-based formula that contains no galactose should be substituted into the infant’s diet.
- Dietary restrictions. Infants with galactosemia must remain on a restricted diet and maintain low blood galactose levels throughout life.
- Read food labels. Labels on all processed foods must be read carefully for ingredients that are milk products.
- Client education. Ascertain the level of understanding of the family caregivers and provide complete information on the disorder.
Evaluation
Goals are met as evidenced by:
- The caregivers identified the appropriate food for the infant.
- The caregivers provided galactose-free milk as a substitution for breast milk.
- The caregivers understood the disease process and the care of the newborn with galactosemia.
Documentation Guidelines
Documentation guidelines for a child with galactosemia include:
- Individual findings include factors affecting, interactions, the nature of social exchanges, and specifics of individual behavior.
- Intake and output.
- Characteristics of stool and vomitus.
- Cultural and religious beliefs, and expectations.
- Plan of care.
- Teaching plan.
- Responses to interventions, teaching, and actions performed.
- Attainment or progress toward the desired outcome.



























Leave a Comment