
Phenylketonuria (PKU) is a rare but critical metabolic disorder that is caused by a deficiency of an enzyme responsible for breaking down the amino acid phenylalanine, leading to its toxic buildup in the body. If left untreated, elevated phenylalanine levels can cause severe cognitive and neurological impairments.
Nurses play an essential role in early detection through newborn screening, supporting families in managing the strict phenylalanine-restricted diet, and monitoring phenylalanine levels to prevent complications.
What is Phenylketonuria?
- Phenylketonuria (PKU), less commonly known as phenylalanine hydroxylase deficiency, is the most common inborn error of amino acid metabolism.
- Phenylketonuria is a recessive hereditary defect of metabolism that, if untreated, causes severe intellectual disability in most but not all affected children.
- It results from an impaired ability to metabolize the essential amino acid phenylalanine, leading to accumulation in blood and tissues.
- Commonly, classic PKU is considered to be present when untreated plasma phenylalanine levels exceed 20 mg/dL (1200 µmol/L) without treatment.
- Elevated phenylalanine levels negatively impact cognitive function, and individuals with classic PKU almost always have an intellectual disability unless levels are controlled through dietary or pharmacologic treatment.
- In the United States and many other countries, PKU is detected by newborn screening, and individuals who are appropriately treated (e.g., with a diet low in phenylalanine and/or tetrahydrobiopterin) can have normal intelligence and lead a normal life.
Pathophysiology
In most patients, the classic type of PKU involves a deficiency of PAH that leads to increased levels of phenylalanine in the plasma (>1200 µmol/L; reference range, 35-90 µmol/L) and to excretion of phenyl pyruvic acid (approximately 1 g/d) and phenylacetic acid in the urine.
- PAH catalyzes the conversion of L-phenylalanine to L-tyrosine, the rate-limiting step in the oxidative degradation of phenylalanine.
- The enzyme PAH crystallizes as a tetramer, with each monomer consisting of a catalytic domain and a tetramerization domain.
- Examination of the mutations causing PKU reveals that some of the most frequent mutations are located at the interface of the catalytic and tetramerization domains.
- A small percentage of children with elevated phenylalanine levels exhibit normal PAH levels but have a deficiency in the synthesis or recycling of BH4.
- This condition is sometimes termed malignant phenylketonuria (PKU) and can result from biallelic mutations in the GCH1, PCB1, PTS, or QDPR genes.
- Thus, individuals with BH4 cofactor deficiency can have additional neurologic problems that are not fully corrected by dietary phenylalanine reduction alone, but often require additional treatments that may not be fully effective.
Statistics and Incidences
PKU frequency varies by population.
- The prevalence in the general US population is approximately 4 cases per 100,000 individuals, and the incidence is 350 cases per million live births.
- Approximately 0.04-1% of the residents in intellectual disability clinics are affected by PKU. A low incidence is reported in African Americans (1/50,000).
- A high incidence is reported in Turkey (approximately 1 case in 2600 births), the Yemenite Jewish population (1/5300), Scotland (1:5300), Estonia (1:8090), Hungary (1/11,000), Denmark (1/12,000), France (1/13,500), the United Kingdom (1/14,300), Norway (1/14,500), China (1/17,000), Italy (1/17,000), Canada (1/20,000), Minas Gerais State in Brazil (1/20,000), and the former Yugoslavia (1/25,042).
- A low incidence is reported in Finland (<1/100,000) and Japan (1/125,000).
- PKU is most commonly diagnosed in neonates because of newborn screening programs.
- Women with PKU must restrict their phenylalanine levels during pregnancy to avoid birth defects and intellectual disability in their infants. Untreated PKU during pregnancy can result in maternal PKU syndrome, which can variably cause congenital heart defects, brain malformations, microcephaly, and intellectual impairment.
- In the United States, PKU is most common in whites; worldwide, PKU is most common in whites and Asians.
Clinical Manifestations
Most individuals with phenylketonuria (PKU) appear normal at birth.
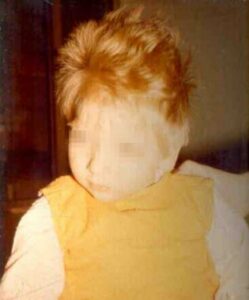
- Fair skin and hair. This is the most characteristic skin manifestation, resulting from impairment of melanin synthesis; it can be striking in black and Japanese patients, although not all untreated patients are fair, and treated patients often have typical pigmentation
- Eczema. Eczema is common, particularly in the perineal area.
- Intellectual disability (ID). Severe, progressive retardation is characteristic.
- Musty odor. There is a characteristic musty smell to the urine.
- Epilepsy. Convulsions may also occur.
- Eye abnormalities. Hypopigmentation may occur in untreated PKU.
Assessment and Diagnostic Findings
Most states require newborns to undergo a blood test to detect the phenylalanine level.
- Guthrie inhibition assay test. This screening uses blood from a simple heel prick; the test is most reliable after the newborn has ingested some form of protein; the accepted practice is to perform the test on the second or third day of life.
- Plasma phenylalanine. A qualified laboratory should measure plasma phenylalanine and tyrosine; screening for PKU includes the determination of phenylalanine levels, the standard amino acid analysis done by means of ion exchange chromatography, or tandem mass spectrometry.
- Urine tests. Results of urine tests (ie, ferric chloride test) may be negative in the first month of life and are rarely used in current practice.
- Magnetic resonance imaging. Cranial MRI studies may be indicated in older individuals who have poor dietary control and are experiencing deficits in motor or cognitive function, or when there are behavioral, cognitive, or psychiatric concerns.
Medical Management
Dietary treatment is required; treatment consists of dietary restriction of phenylalanine often with tyrosine supplementation.
- Phenylalanine-free formulas. A formula low in phenylalanine should be started as soon as the condition is detected; Lofenalac and Phenyl-free are low phenylalanine formulas; best results are obtained if the special formula is started before the newborn is 3 weeks of age.
- Restricted diet. A low phenylalanine diet is a very restricted one; foods to be omitted are bread, meat, fish, dairy products, nuts, and legumes; the child remains on the diet at least into early adulthood, and it may even be recommended indefinitely.
- Amino acid supplementation. Other essential amino acids are supplemented using various medical foods, and vitamin, mineral, and other micronutrients are followed closely; stringent phenylalanine-restricted diets have been reported to cause deficiencies of iron, zinc, selenium, and other nutrients and essential amino acids in patients with PKU.
- Avoidance of aspartame. Aspartame must also be avoided; phenylalanine is one of the primary components of aspartame; it is found in many artificially sweetened foods and soft drinks, as well as some vitamins and medicines; a 12-oz can of aspartame-sweetened diet drink contains approximately 105 mg of phenylalanine (ie, 25-50% of the usual daily intake).
Pharmacologic Management
Patients who refuse dietary treatment may benefit to some degree from consuming large neutral amino acids.
- Sapropterin. Some patients with PKU experience a significant lowering of plasma phenylalanine levels after administration of sapropterin, a commercially available, FDA-approved form of the tetrahydrobiopterin (BH4) cofactor.
- Enzyme therapy. An alternative enzyme therapy for PKU in clinical trials involves the use of an injectable form of phenylalanine ammonium lyase, an enzyme capable of substituting for phenylalanine hydroxylase (PAH); this therapy is currently under investigation for the potential treatment of patients with PKU who do not respond to BH4.
Nursing Management
Nursing care for a child with PKU involves the following:
Nursing Assessment
Assessing a child with PKU should include:
- Nutritional history. Upon the birth of the newborn with PKU, assess if he has consumed any formula which is not phenylalanine-free to identify measures to be made.
- Physical examination. Assess the newborn for any manifestations that may indicate emergency care.
Nursing Diagnoses
Based on the assessment data, the major nursing diagnoses are:
- Imbalanced nutrition: less than body requirements related to a restrictive diet.
- Impaired skin integrity related to scratching at the perineal area secondary to eczema.
- Risk for injury related to convulsions.
- Anxiety related to the disorder and adverse reactions that the infant may experience.
- Altered thought processes related to poor intellectual abilities.
- Deficient knowledge of caregivers related to the disorder and the care of the newborn with PKU.
Nursing Care Planning and Goals
The nursing care planning goals for a child with phenylketonuria are:
- The caregiver will be able to provide the appropriate nutritional needs of the infant.
- The infant will be free from injury.
- The infant’s skin integrity will be intact and free from infection.
- The caregiver will be knowledgeable enough about the disorder and in managing the infant with PKU.
Nursing Interventions
The nursing interventions for a child with PKU are:
- Diet. Inform family caregivers of the foods that they should avoid giving the infant once he is allowed to take solid foods; special formulas are also indicated instead of regular formulas or breastmilk, such as Lofenalac and Phenyl-free formulas.
- Emotional support. Offer support to the family emotionally especially after the diagnosis so they could cope with the shock, anxiety, and stress.
- Health education. Educate the family on the disease process and how they could help the child grow as normally as he could; if a child’s phenylalanine control is kept within the acceptable range, growth, and development will not be affected.
- Guidance from a dietitian. It is important that dietary advice is consistent; the child’s dietitian should, therefore, be the only person giving advice.
- Safety. In case of convulsions which may occur in a child with PKU, educate the family caregivers on how to handle the child with safety being the priority.
Evaluation
Goals are met as evidenced by:
- The caregiver provided the appropriate nutritional needs of the infant.
- The infant is free from injury.
- The infant’s skin integrity is intact and free from infection.
- The caregiver is knowledgeable enough about the disorder and in managing the infant with PKU.
Documentation Guidelines
Documentation in a child with PKU includes:
- Individual findings include factors affecting, interactions, the nature of social exchanges, and specifics of individual behavior.
- Intake and output.
- Cultural and religious beliefs, and expectations.
- Plan of care.
- Teaching plan.
- Responses to interventions, teaching, and actions performed.
- Attainment or progress toward the desired outcome.


























Leave a Comment